Genome Organization and Reorganization in Evolution: Formatting for Computation and Function
James A. Shapiroa
Department of Biochemistry and Molecular Biology
University of Chicago
920 E. 58th Street, Chicago, IL 60637, USA
aTEL:773-702-1625, FAX 773-702-0439; EMAIL: jsha@midway.uchicago.edu
KEY WORDS: evolution, genome formatting, genome system, system architecture, natural genetic engineering, computation, signal transduction, DNA rearrangements, information storage, repetitive DNA
Presented at a symposium on "Contextualizing the Genome," Ghent University,
Belgium, November 25 - 28, 2001
(Ann. N.Y. Acad. Sci., in press)
Abstract:
This volume deals with the role of Epigenetics in life and evolution.
The most dynamic forms of functional genome formatting involve DNA interacting
with cellular complexes that do not alter sequence information. Such important
epigenetic phenomena are the main subjects of other articles in this volume.
This article focuses on the long-lived form of genome formatting that lies
within the DNA sequence itself. I argue for a computational view of genome
function as the long-term information storage organelle of each cell. Structural
formatting consists of organizing various signals and coding sequences
into computationally-ready systems facilitating genome expression and genome
transmission. The basic features of genome organization can be understood
by examining the E. coli lac operon as a paradigmatic genomic system.
Multiple systems are connected through distributed signals and repetitive
DNA to form higher-order Genome System Architectures. Molecular discoveries
about mechanisms of DNA restructuring show that cells possess the Natural
Genetic Engineering functions necessary for evolutionary change by rearranging
genomic components and reorganizing system architectures. The concepts
of cellular computation and decision-making, genome system architecture,
and natural genetic engineering combine to provide a new way of framing
evolutionary theories and understanding genome sequence information.
INTRODUCTION: Conceptual Shifts at the Turn of the Century
The symposium on "Contextualizing the Genome" comes at the start of
a new century and at a key period in the study of heredity and evolution.
The 20th Century began with the rediscovery of Mendelism and
has been called "The Century of the Gene." The 21st Century
has begun with the publication of the draft Human Genome sequence and is
quite likely to be called "The Century of the Genome." The genome comprises
all the DNA sequence information of a particular cell, organism or species.
Reading the genome has been a major goal of molecular biologists since
the 1953 discovery of the double-helical structure of DNA. I will argue
in this article that what seems like a modest change in terminology from
"gene" to "genome" actually reflects a tremendous advance in knowledge
and a profound shift in the basic concepts behind our thinking about the
workings of living cells (Table 1).
| Table 1. Conceptual changes resulting from molecular biology discoveries | ||||
| Conceptual Category | 20th "Century of the Gene" | 21st "Century of the Genome" | ||
| Dominant scientific perspective | Reductionism | Complex Systems | ||
| Fundamental mode of biological operation | Mechanical | Cybernetic | ||
| Central focus of hereditary theory | Genes as units of inheritance and function | Genomes as interactive information systems | ||
| Genome organization metaphor | Beads on a string | Computer operating system | ||
| Sources of inherited novelty | Localized mutations altering one gene at a time due to physico-chemical insults or replication errors | Epigenetic modifications and rearrangement of genomic subsystems by internal natural genetic engineering functions | ||
| Evolutionary processes | Background random mutation and natural selection of small increases in fitness; cells passive | Crisis-induced, non-random, genome-wide rearrangements leading to novel genome system architectures; cells actively engineering their DNA | ||
There is a fine irony in the conceptual changes summarized in Table 1. The expectation of its pioneers was that molecular biology would confirm the reductionist, mechanical view of life.13 However, the actual result of molecular studies of heredity, cell biology and multicellular development has been to reveal a realm of sensitivity, communication, computation and indescribable complexity.46 This years Nobel Prize in Medicine illustrates this point: the recipients were recognized for identifying components of the molecular computational network that regulates the eukaryotic cell cycle.7 Special mention was made of the concept of checkpoints, the inherently computational idea that cells monitor their own internal processes and make decisions about whether to proceed with the various steps in cell division based on the information detected by surveillance networks.
In addition to uncovering intra- and inter-cellular computing systems (frequently referred to as "signal transduction" networks), molecular analysis has also confirmed the generality of Barbara McClintocks revolutionary discoveries of internal systems for genome repair and genome restructuring.8 The ability of all living cells to take action to conserve or change their DNA sequence information was unknown when the basic concepts of Mendelian genetics were formulated. In that period of ignorance, it was assumed that genomes are constant and only change by accident. The discovery of repair systems, mutator functions and mobile genetic elements (MGEs) brought the phenomena of mutation out of the realm of stochastic processes and into the realm of cellular biochemistry.9-15 DNA biochemistry is not fundamentally different from the biochemistry of metabolism or morphogenesis. Consequently, our notions about the evolutionary sources of genomic differences that underlie biological diversity and adaptive specialization require a profound re-evaluation. All aspects of cellular biochemistry are subject to computational regulation. So we can no longer make the simplifying assumption of randomness, and we have to incorporate the potential for biological specificity and feedback into evolutionary thinking.
THE GENOME IN CONTEXT: Where Does the Genome Fit in the Information Economy of the Cell ?
If we wish to place the genome in context, we need
to demystify DNA and cease to consider it the complete "blueprint of life."
The genome serves as the long-term information storage organelle of each
living cell. It contains several different classes of information each
involving a particular kind of DNA sequence code (Table 2).16
| Table 2. Different Classes of Information Stored in Genome Sequence Codes |
| Coding sequences for RNA and protein molecules |
| Identifiers for groups of coding sequences expressed coordinately or sequentially |
| Sites for initiating and terminating transcription of DNA into RNA |
| Signals for processing primary transcripts to smaller functional RNAs |
| Control sequences setting the appropriate level of expression under specific conditions |
| Sequence determinants marking domains for chromatin condensation and chromatin remodeling |
| Binding sites affecting spatial organization of the genome in the nucleus or nucleoid |
| Sites for covalent modification of the DNA (such as methylation) |
| Control sequences for initiating DNA
replication
Sequence structures permitting complete replication at the ends of linear DNA molecules (telomeres) |
| Centromeres and partitioning sites for equal distribution of duplicated DNA molecules to daughter cells following cell division (non-random chromosome partitioning) |
| Signals for error correction and damage repair |
| Sites for genome reorganization (DNA rearrangements) |
The best current metaphor for how the genome operates
is to compare it to the hard drive in an electronic information system
and think of DNA as a data storage medium. The metaphor is not exact, in
part because genomes replicate and are transmitted to progeny cells in
ways that have no precise electronic parallel. Nonetheless, the information-processing
metaphor allows us to view the role of the genome in a realistic context.
DNA by itself is inert. Information stored in genomic sequences can only
achieve functional expression through interaction of DNA with other cellular
information systems (Table 3).
| Table 3. Functional interactions between the genome and other cellular information systems | ||
| Information System | Function | |
| DNA replication | Duplicate the genome | |
| Chromosome segregation | Transmit a complete genome to each daughter cell | |
| Basic transcription | Copy DNA into RNA | |
| Transcription factors and signal transduction networks | Control timing and level of transcription, establish differential expression patterns | |
| DNA compaction (chromatin modeling) | Control accessibility of genome regions, often comprising many loci; maintain differentiation | |
| Covalent DNA modification (e.g. methylation) | Control chromatin formatting, interactions with transcription apparatus | |
| Natural genetic engineering | Create novel DNA sequence information | |
As I will argue shortly in more detail, the molecular interactions relating to genome function are intrinsically computational (i.e. they involve multiple inputs that need to be evaluated algorithmically to generate the appropriate cellular outcome). Since functional information can only be extracted from the genome by computational interactions, organismal characteristics (phenotypic traits) are not necessarily hard-wired in the DNA sequence. There is no linear genotype-phenotype relationship. In organisms with complex life cycles, for example, the same genome encodes the morphogenesis of quite distinct creatures at different developmental stages (e.g. caterpillars and butterflies). Within species ranging from bacteria to higher plants and animals, differentiated cell types share the same genome but express alternative sets of coding information. Moreover, individuals of the same species can have markedly different morphologies in distinct environments or at different times of the year.17
If we reflect on the immense complexity of cellular activity as revealed by modern biochemistry and cell biology, we can appreciate the need for constant monitoring, computation and decision-making to keep millions of molecular events and chemical reactions from undergoing chaotic transitions and spinning out of control. Chromosome distribution at eukaryotic mitotic cell division provides a good illustration of the communication/decision-making control principle.5,18, 19 By ensuring that each daughter cell receives one and only one homologue copy of each duplicated chromosome, this is a highly non-random process. (If n chromosomes duplicated and then segregated into daughter cells randomly, the chance of each daughter receiving a full complement would be 2-n.) Equal distribution is guaranteed by a checkpoint system delaying the active phase of cell separation (cytokinesis) until the duplicated and paired homologues are aligned along the metaphase plate and attached by microtubules to opposite spindle poles. Proper alignment and spindle pole attachment then lead to distribution of one homologue to each daughter cell at cytokinesis. Chromosome pairs that are not properly aligned and attached emit chemical signals. These signals are interpreted by the cell cycle control network and the homologue separation machinery as "WAIT" messages. In this way, the dynamic process of microtubules searching to attach onto unbound homologues is allowed to continue to completion. Only then, when every chromosome pair experiences the appropriate mechanical tension, does the inhibitory signal disappear and the cell make the decision to begin the series of events that separate the chromosomes and form two daughter cells.
Applying the computer storage system metaphor, the ideas summarized in Tables 2 and 3 can be restated by saying that the genome is formatted for interaction with cellular complexes that operate to replicate, transmit, read, package and reorganize DNA sequence information. Genome formatting is similar to the formatting of computer programs in that a variety of generic signals are assigned to identify files independently of their unique data content. We know that different computer systems employ different signals and architectures to retrieve data and execute programs. In an analogous fashion, diverse taxonomic groups often employ characteristic DNA sequences and chromosomal structures to organize coding information and to format their genomes for expression and transmission. Thus, one of the consequences of evolutionary diversification is the elaboration of distinct Genome System Architectures.20
The natural genetic engineering system has the job of restructuring the genome (Table 3). The presence of genomic rewriting functions makes very good sense in terms of the idea that DNA is a data storage medium. Clearly, a medium in which new data and new programs can be written is far more valuable than a read-only memory device. Reverse transcription, for example, is a way of storing data in the genome about transcriptional and RNA processing events.21,22 Such stored data can later be accessed and incorporated into new genetic structures by DNA rearrangement activities. In this way, natural genetic engineering facilitates evolutionary success.23
SYSTEMS ORGANIZATION OF GENOMIC INFORMATION: Deconstructing the gene, combinatorial structure of genomic determinants, and the computational nature of regulatory decisions.
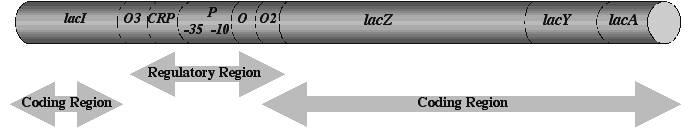
A good way to appreciate the conceptual changes resulting from molecular studies of the genome is to examine the history of a paradigmatic genetic locus, the E. coli lac operon.24-26 Like all classically defined "genes," the lac operon began existence as a single point on a genetic map, denoting the location of mutations affecting the ability of E. coli cells to utilize the sugar lactose. The lac operon is a paradigm because molecular genetic analysis of this locus led to our current ideas about how cells regulate the expression of protein-coding information in DNA. It is significant that lac posed a problem in cellular perception and adaptation. In his doctoral thesis, Monod27 had discovered that E. coli cells could distinguish between glucose and lactose in a mixture of the two sugars; the bacteria consumed all available glucose before digesting the lactose. Monod and his colleagues spent the next two decades elucidating how E. coli cells accomplish this discrimination (i.e. adjust their metabolism to use one sugar before the other). They found that the lac "gene" resolved itself into four different coding regions plus a completely new class of genetic determinant, a DNA site where regulatory molecules bind and control the reading of adjacent DNA sequences.24, 28 Subsequent research identified further control sites so that by the 1990s, the lac operon could be schematized as in Figure 1.
Figure 1. The lac operon about 1990 (not to scale). The genetic designations for each determinant (in italics) indicate the following functional roles: lacI = coding sequence for the repressor molecule; lacO, O2, O3 = operator sequences, binding sites for dimers of LacI repressor; CRP = binding site for the complex of cyclic AMP (cAMP) plus CRP (the cAMP Receptor Protein that stabilizes RNA polymerase binding to lacP; lacP = promoter sequence, binding site for RNA polymerase to initiate transcription, composed of distinct 10, -35 binding sites; lacZ = coding sequence for beta-galactosidase enzyme (major reaction: hydrolyzes lactose, minor reaction: converts lactose to allolactose, the inducer that binds repressor); lacY = coding sequence for lactose permease (actively transports lactose into cell); lacA = coding sequence for galactoside transacetylase (acetylates toxic lactose analogues).
Molecular dissection had transformed the dimensionless lac "gene" into a system composed of regulatory sites and coding sequences. The atomistic term "gene" no longer adequately describes such a tightly linked genomic system, and the less conceptually loaded term "genetic locus" is more appropriate. The importance of identifying lacO, lacP, and CRP cannot be overemphasized. These and other binding sites in DNA are not "genes" in any classical sense of the term. They do not encode the synthesis of a specific product. Rather, they constitute signals formatting the DNA for transcription. While some binding sites are quite specific, such as the operators that are only found in the lac operon, most are generic and can be found associated with multiple coding sequences or in multiple genomic locations. CRP sites, for example, format a series of catabolic operons in the E. coli for common regulation by glucose,29 while lacP belongs to a family of promoter sites that enable transcription during active growth conditions.30 Such distributed protein binding sites in DNA are central to our understanding of how various cellular information systems interact with the genome (Table 3).5
The computation-enabling aspects of lac operon organization become apparent when we understand how the various regulatory sites connect this locus to physiological data about glucose and lactose metabolism. The cell senses the presence of glucose indirectly by means of its uptake system.29 When glucose is available, a membrane-associated protein involved in transporting the sugar into the cell continually transfers phosphate groups to the sugar molecule, which enters the cell in a phosphorylated form. The transport protein itself thus exists almost all the time in the unphosphorylated form. When glucose is no longer available, this protein has no acceptor for its phosphate groups and so exists continuously in the phosphorylated form. When phosphorylated, it acquires the ability to activate the enzyme adenylate cyclase, which converts ATP into cAMP, thus raising the intracellular concentration of cAMP. The cell uses the phosphorylated transport protein and a high cAMP concentration as indicators that glucose is not available. The cAMP concentration is read by the CRP protein which binds to the CRP site in lac only in the presence of abundant cAMP. The presence of the cAMP-CRP complex bound to lac DNA stabilizes the contacts between lacP and RNA polymerase and so informs the transcription apparatus that the lac operon is ready for transcription. In the absence of lactose, however, only rare transcription events can occur because LacI repressor molecules bind to two of the operator sites and create a loop in the DNA, blocking access to the lacP promoter. The cell also senses the presence of lactose indirectly. Low levels of LacY permease transport a few lactose molecules into the cell, where LacZ beta-galactosidase converts some of them to a related sugar called allolactose. Allolactose can bind to LacI repressor, induce a change in shape that makes the repressor unable to bind lacO, and so free lacP for transcription. Each of these molecular interactions constitutes an information transfer event, or logical statement, and the combination of all of them allows the bacterial cell to compute the algorithm enabling discrimination between the two sugars: "TRANSCRIBE lacZYA IF AND ONLY IF GLUCOSE IS NOT PRESENT, LACTOSE IS PRESENT, AND THE CELL CAN SYNTHESIZE FUNCTIONAL PERMEASE AND BETA-GALACTOSIDASE". 26
Two features of the lac operon regulatory computation are particularly noteworthy and generalizable. (1) Information transfer occurs by the use of chemical symbols to represent empirical data about the physiological environment: cAMP, allolactose and protein phosphorylation levels represent the availability of glucose and lactose. (2) The regulatory network integrates many different aspects of cell activity (transport, cytoplasmic enzymology and energy metabolism) into the transcriptional decision. In other words, it is literally impossible to separate physiology from genomic regulation in E. coli and, indeed, in any living cells.5, 6
HIERARCHIES IN GENOME FORMATTING: Multiple levels of combining genomic determinants, chromatin formatting, repetitive DNA and Genome System Architecture
The systems view of genomic organization applies at all levels. The lowest level genomic determinants, such as protein binding sites, themselves consist of multiple interacting components. For example, lacO and CRP are each DNA palindromes, consisting of head-to-head repeats of the same short sequence, thereby permitting the cooperative binding of two LacI repressor or CRP subunits in dimeric protein structures.29, 30 Likewise, the lacP site actually consists of two subsites which must be separated by 16 or 17 base pairs for proper RNA polymerase binding.30 Even protein coding sequences are systems. In eukaryotes, of course, they are often broken up into separate exons, which must be spliced together in the messenger RNA to construct an active coding sequence, and we now appreciate how important regulation of the splicing process is in contributing to controlled production of different proteins from a single primary transcript.31 But in all organisms, even bacteria where there are almost no introns, we now view proteins and their coding sequences as systems of interacting domains.32 For example, the LacI repressor molecule has separate domains for DNA binding, for protein-protein binding and for binding the allolactose inducer. As genome sequencing shows, most major steps in protein evolution occur by forming new combinations of domains, a process involving both domain swapping and domain accretion.33
At higher levels, the metabolic and developmental regulatory circuits that control cell physiology, cell differentiation, morphogenesis and multicellular development are based on the combinatorial principle of arranging specific binding sites so that the proteins and DNA can interact in ways that allow the cell to process molecular information and compute whether to transcribe particular coding sequences.5, 6 Common binding sites serve to connect different genetic loci into coordinated expression systems, and various combinations of sites interact to execute far more sophisticated decisions than the one described above.34, 35
Cases where functioning of large genomic regions, often comprising multiple genetic loci, come under cellular control are particularly relevant to this Symposium.36 The way the genome is compacted into the DNA-protein complex known as chromatin has a profound influence on the interactions summarized in Table 3. By differential compaction, cells can place long stretches of individual chromosomes into active or inactive chromatin domains. This mode of genome regulation is considered to be "epigenetic."37 Cells use chromatin formatting to execute complex programmatic tasks, such as expressing developmentally-specific homeobox proteins in precise patterns along the animal body axis.38 Like transcriptional regulation of individual loci, chromatin formatting depends on certain kinds of dispersed binding sites and small determinants, such as the "insulator elements" that form the boundaries between distinct chromatin domains.39
Chromatin formatting also involves the important (yet often dismissed) class of genomic determinants known as "repetitive DNA sequences." Repetitive sequences can vary in length from a few up to thousands of base-pairs, and they can be present at frequencies that range from only 2 or 3 copies up to hundreds of thousands of copies per haploid genome.40 In the human genome, for example, repetitive sequences comprise well over 50% of the total DNA (compared to less than 5% for protein-coding exons).33 Repetitive elements influence chromatin structure in two ways. Dispersed repeat copies (Figure 2) may contain binding sites for chromatin organizing proteins, so that they form part of the genetic basis for local chromatin structure. But a more general influence occurs with tandem head-to-tail arrays of a single repetitive sequence (Figure 2). As these arrays grow longer, they tend to nucleate the formation of a highly compacted structure called "heterochromatin." 41 Heterochromatin inhibits transcription and recombination and delays replication, generally blocking expression of coding sequence information.

Figure 2. Dispersed and tandem arrangements of repetitive DNA sequences.
Regions of heterochromatin can spread along chromosomes. Thus, the presence of a region containing tandem repeats can nucleate a heterochromatic domain and negatively affect the expression of genetic loci at distances of many kilobase-pairs. This so-called "position effect" phenomenon is well-known in fruit flies, where chromosome rearrangements can inhibit visible characters (such as eye pigmentation) by placing loci encoding proteins needed for expression of those characters near heterochromatin blocks at centromeres.42 Position effect is not limited to visible phenotypes. Analogous rearrangements also lead to loss of essential functions, and the same genetic backgrounds that suppress position effect on visible phenotypes also relieve lethality. 42
The position effect phenomenon provides a very direct demonstration that the genome is a large system integrated in part by its content of repetitive DNA. By altering dosage of the largely heterochromatic Y chromosome, fruit fly geneticists can alter the total amount of tandem repetitive DNA in the genome.41, 42 When they increase the amount of heterochromatin in XYY males, the inhibition on expression of a rearranged eye pigmentation locus is reduced, presumably because the extra repetitive DNA binds and titrates proteins needed to form heterochromatic domains. When total heterochromatin decreases in XO males, the inhibition becomes more severe, as expected. Alteration of heterochromatin-specific DNA binding protein levels has just the opposite effects: loss of these proteins relieves or suppresses position effect, while overexpression enhances it.43 Since suppression or enhancement of position effect occurs when the bulk of genomic heterochromatin is located on a different chromosome from the inhibited locus, it is clear that repetitive DNA can act both in cis and in trans to influence the epigenetic formatting of genetic loci.
In addition to influencing chromatin organization and expression, repetitive sequences play a number of important roles in genome transmission. For example, they are involved in forming centromeres, the sites where chromosomes attach to microtubules for separation at cell division,44 in replicating the ends of linear chromosomes,45 and in chromosome pairing during the formation of gametes.46 We have sufficient current knowledge to state definitively that the distribution of repetitive DNA sequence elements is a key determinant of howa particular genome functions (i.e. replicates, transmits to future generations, and encodes phenotypic traits). Including distributed protein binding sites as repetitive elements, it is clear that repetitive DNA formats coding sequences and genome maintenance routines in the same way that generic digital signals format individual data files and programs for use by a particular computer system architecture. In other words, each genome has a characteristic Genome System Architecturethat depends in large measure on its repetitive DNA content.
EVOLUTIONARY IMPLICATIONS OF GENOME SYSTEM ARCHITECTURE: NATURAL GENETIC ENGINEERING
A key aspect of evolution is the emergence of new genome structures carrying the information necessary for the epigenesis of new organismal phenotypes. According to the principles just outlined, genomic novelties may arise by two processes:
| Table 4. Natural Genetic Engineering Capabilities | |
| DNA Reorganization Functions | DNA Rearrangements Carried Out |
| Homologous recombination systems49 | Reciprocal exchange (homologous crossing-over); amplification or reduction of tandem arrays (unequal crossing-over); duplication, deletion, inversion or transposition of segments flanked by dispersed repeats; gene conversion |
| Site-specific recombination50 | Insertion, deletion or inversion of DNA carrying specific sites; serial events to build operons, tandem arrays |
| Site-specific DNA cleavage functions | Direct localized gene conversion by homologous recombination47; create substrates for gene fusions by NHEJ (VDJ recombination in the immune system52-54) |
| Non-homologous end-joining (NHEJ) systems55 | Precise and imprecise joining of broken DNA ends; create genetic fusions; facilitate localized hypermutation54 |
| Mutator DNA polymerases56 | Localized hypermutation |
| DNA transposons10-15 | Self insertion, excision; carry signals for transcriptional control, RNA splicing and DNA bending; non-homologous rearrangements of adjacent DNA sequences (deletion, inversion or mobilization to new genomic locations); amplifications |
| Retroviruses and other terminally repeated retrotransposons10-15 | Self insertion and amplification; carry signals for transcriptional control, RNA splicing and chromatin formatting; mobilization of sequences acquired from other cellular RNAs |
| Retrotransposons without terminal repeats10-15, 57 | Self insertion and amplification; carry signals for transcriptional control and RNA splicing; reverse transcription of cellular RNAs and insertion of the cDNA copies; amplification and dispersal of intron-free coding sequences; mobilization of adjacent DNA to new locations (e.g. exon shuffling58) |
| Terminal transferases | Extend DNA ends for NHEJ; create new (i.e. untemplated) DNA sequences in the genome52 |
| Telomerases59 | Extend DNA ends for replication |
When we look carefully in experimental situations, we find that the vast majority of genetic changes, even the point mutations previously ascribed to stochastic causes, result from the action of natural genetic engineering functions. The accidents are efficiently removed by cellular proofreading and repair systems.9 The fact that the sources of DNA sequence variability are internal and biochemical has a number of implications for how we make assumptions about the genetic aspects of evolution. First, we no longer need to think of change as small and localized. Natural genetic engineering functions can fuse and rearrange distant regions of the genome, and many of the changes involve large segments of DNA (e.g. insertion of a cDNA copy kilobase-pairs in length or translocation of a chromosome segment measuring many megabase-pairs). Secondly, each change is not necessarily independent of other changes. A natural genetic engineering system, once active, can mediate more than one DNA rearrangement event, and a single event can produce a cluster of changes (e.g. multiple base substitutions resulting from localized hypermutation). Third, the changes are not random in nature. Each kind of natural genetic engineering function (Table 4) acts on the DNA in specific ways and usually displays characteristic affinities for DNA sequence and chromatin structure. The movement of a particular MGE into different genomic locations is inherently non-random because each insertion event carries the same set of regulatory, cleavage and coding sequences to the new location. Moreover, most MGEs display a significant degree of "hotspotting" in their insertions, and the action of even general systems, like homologous recombination51 or NHEJ55, can be targeted by site-specific DNA cleavage activities, as it is in immune system rearrangements and hypermutation.53,54
There is abundant evidence that internal genetic engineering systems have been major actors in natural populations and in genome evolution. Our own survival literally depends upon genetic engineering. Our immune system cells form an essentially infinite array of antigen recognition molecules by rearranging and specifically mutating the corresponding DNA sequences.52-54 In some organisms, genome restructuring is part of the normal life cycle. In the ciliated protozoa, for example, the germ line genome is regularly fragmented into hundreds of thousands of segments, which are then processed and correctly reassembled to create a functioning somatic genome of radically different system architecture.60 43% of the human genome, for example, consists of MGEs,33 and hundreds of thousands of retrotransposons (SINE elements) characterize the genomes of each mammalian order.61 Evolution of mammalian genomes has thus involved literally >100,000 transposition and retrotransposition events. In certain well-studied groups of organisms, such as natural fruit fly populations, we can now identify MGEs that produce the chromosome rearrangements that distinguish different species.62 Genome sequencing has provided numerous examples of "segmental duplications" in higher plants and animal genomes.33, 47, 48 These duplications involve the kinds of chromosome segment movements made possible by natural genetic engineering processes (Table 4, Figure 3). In addition, coding sequence amplifications have produced so-called "gene families" in most genomes. In the human genome, the large family encoding olfactory receptor proteins is composed mainly of intron-free copies and apparently evolved from multiple retrotransposition events.63 Finally, we are beginning to obtain direct evidence for the participation of MGEs in the evolution of regulatory regions64-66 and protein coding sequences.64, 65, 67 A particularly instructive example is the sequence encoding a rodent ion channel (Figure 3). More than half this coding sequence derives from rodent-specific SINEs, making it a sequence that could only have evolved in rodents and not in other kinds of mammals.67
From the foregoing, it is evident that the capacity of living cells to carry out massive, non-random, genome-wide DNA rearrangements has to be incorporated into any theory of evolutionary change. If we pause to reflect that every existing organism is a survivor of an evolutionary process involving multiple possibilities of extinction, then the power of natural genetic engineering should not surprise us. Organisms that can create useful genomic novelty most rapidly and effectively will have the best chance of surviving an evolutionary crisis. Indeed, the hardest proposition to accept is the assertion that organisms have not optimized their ability to expand and rewrite the information stored in their genomes. A species that depends exclusively on independent random changes for inherited novelty will not be very competitive in the evolutionary sweepstakes.
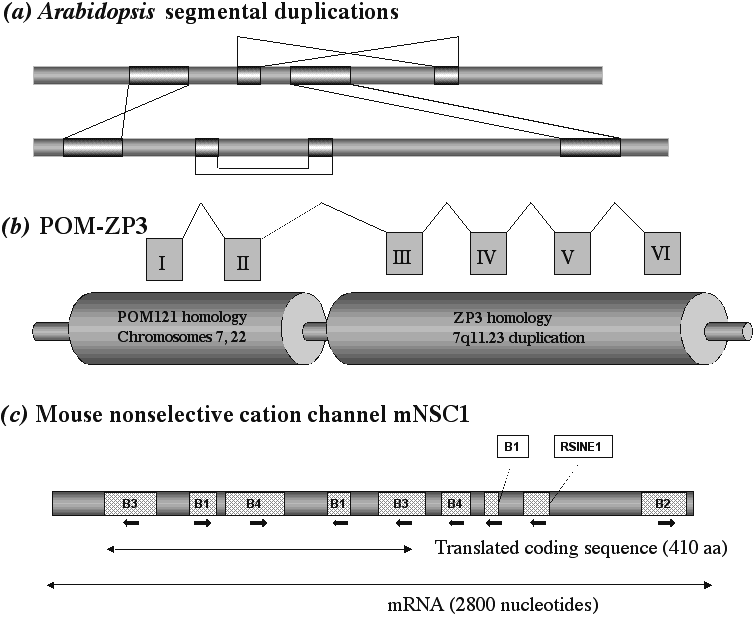
Figure 3. Natural Genetic Engineering Products in Sequenced Genomes. (a) The kind of large segmental duplications observed in the Arabidopsis thaliana genome. The patterned rectangles illustrate segments several Mbp long that are duplicated either within one chromosome or between chromosomes. Crossed lines indicate the orientation has been inverted.47 (b)A hybrid transcription unit resulting from segmental duplication in the human genome.48 The1.6-kb POM-ZP3 transcript from chromosome region 7q11.23 is encoded by a chromosome-specific duplication of the ZP3A locus (zona pellucida glyocprotein gene 3A) juxtaposed to two exons of the POM125 (perinuclear outer membrane) locus. Multiple copies of POM125 segmental duplications are found on chromosomes 7 and 22. The fusion transcript encodes a 250 amino acid protein; the first 76 amino acids are 83% identical to rat POM125, and the remaining 124 amino acids are 98% identical to ZP3. (c) The structure of 2800 nucleotide mRNA encoding mouse cation channel protein mNSC1. About half the mRNA sequence and >50% of the protein coding sequence derive from rodent-specific SINE elements.67
CELLULAR REGULATION OF NATURAL GENETIC ENGINEERING: Computational potential in evolution
The most profound conceptual result of learning about natural genetic engineering and epigenetic imprinting is that they place the processes of heritable variation in the realm of cell biology, where events are subject to computational decisions involving biological inputs. By removing variation from the realm of stochastic processes (without making it subject to any kind of rigid determinism), we can begin to think about how the genomic basis of evolutionary change fits into contemporary ideas about life as self-regulating complexity. There are two key areas where we have experimental evidence and even some degree of mechanistic understanding to guide us: (1) the connection between life experience and natural genetic engineering events, and (2) the interaction between the networks governing transcriptional control and chromatin formatting and those governing the choice of genomic targets for natural genetic engineering activities. We know that cells can control natural genetic engineering in response to life history events and direct their activities to specific places in the genome because those abilities are embodied in our immune system: human lymphocytes display both developmental control of DNA rearrangements and mutational specificity.52-54
In her Nobel Prize address, McClintock spoke of genomic reaction to challenge and posed questions about "how the cell senses danger and instigates responses to it that often are truly remarkable".8 McClintock introduced the concept of "genome shock" to encompass those inputs which lead to activation of DNA rearrangement functions, and there is general agreement among biologists that stress leads to increased mutability. In certain carefully-studied systems, we can define both the "shocks" and the molecular circuits that respond to them with greater detail. As McClintock pointed out, the SOS DNA damage response of bacteria is the paradigm genome-monitoring and inducible reaction system. SOS depends upon the ability of the RecA protein to recognize single-stranded gaps in DNA resulting from replication blocks and then inactivate the LexA repressor, which blocks transcription of a number of cellular repair, recombination, checkpoint, mutator polymerase, and programmed cell death functions.68 By layering the various repair routines, by providing differential sensitivity to RecA derepression for each function, and by engaging positive and negative feedback loops on RecA control activities, the SOS system endows the bacterial cell with a sophisticated, modulated response to certain classes of DNA damage, such as double-strand breaks. Eukaryotic cells have a far more complex system that responds to inputs about DNA damage, cell physiology and extracellular growth factors and makes the decision between repair and programmed cell death.69 From studies of tumor cells that acquire mutations affecting components of this response system, we know that breakdown of the control network is involved in the genetic instabilities that lead to malignancy.70 Thus, cancer may be considered a cellular information-processing pathology.
A good example of genome shock is the phenomenon of "adaptive mutation."25, 71 This kind of environmentally-induced genetic change occurs in aerobic starving bacteria under selection. The cells are stimulated to produce many DNA changes, some of which enable them to adapt to selective conditions and recover the ability to proliferate. In the first adaptive mutation system described, a DNA transposon is activated to create a fused protein coding sequence,72 and the activation process includes transcription factors, DNA binding proteins, and regulatory proteases.73 Another well-studied adaptive mutation system examines recombination-dependent lac33 frameshift reversion; in that case, activation involves aerobic response factors and the SOS system.71, 74
The activities of MGEs are subject to a wide range of regulatory routines (including epigenetic control by DNA methylation). From an evolutionary perspective, one of the most important life history events that activates MGEs is hybridization, or mating between individuals of two different populations or species. Hybridization, not selection, is the way that breeders make new species.75 In fruit flies, where the phenomenon has been particularly well studied, germ-line instabilities result from transposable element activation after mating between separate populations. These instabilities include mutations, chromosome breakage, chromosome rearrangements, mobilizations of transposable elements, and female sterility; all these germline disfunctions have been placed under the rubric of "hybrid dysgenesis," and a causative role has been established for both DNA transposons76 and retrotransposons.77, 78
Two features of hybrid dysgenesis make it particularly instructive for potential models of evolutionary change. One feature is that many copies of the responsible MGEs are typically involved, so that the genomes of dysgenic flies undergo many concurrent changes and acquire new organizational properties. The second feature is that these multiple changes occur during the mitotic development of the germ line, so that a cell with a reorganized genome will undergo a number of cell divisions before meiosis and production of gametes. Consequently, a number of offspring from a single dysgenic fly can share novel chromosome configurations. In this way, an interbreeding population with a dramatically reorganized genome can appear in a single generation. Comparable examples of hybrid instabilities have been documented in marsupials and natural mice populations.79, 80 Of particular relevance to this Symposium is the observation that loss of DNA methylation follows hybridization and accompanies activation of retrotransposons in mammals79 and plants.81
Cellular regulatory networks not only control when
genomes undergo reorganization. They also are able to modulate the locations
where natural genetic engineering functions operate (Table 5). In some
cases, we understand at least something about the mechanisms that produce
target choice. The R1 and R2 retrotransposons encode a site-specific endonuclease
that targets their insertion into the 28S ribosomal RNA coding sequence
and similar sequences elsewhere in the arthropod genome.89 Group
II "homing" introns use a similar retrotransposition mechanism,93while
Group I homing introns use a site-specific endonuclease in combination
with homologous recombination to carry out mobility completely at the DNA
level.92 Where control is exercised through chromatin formatting,
it is not hard to see how different chromatin configurations will affect
access to distinct genomic locations by the proteins and nucleic acids
that produce DNA reorganization. But the results are not always intuitively
obvious. For example, the Ty5 retroviral-like element of brewers yeast
has a strong preference for chromatin that has been transcriptionally silenced
and is not open to the transcriptional apparatus.95
| Table 5. Some examples of non-random targeting in natural genetic engineering | |
| DNA reorganization system | Observed specificity |
| Immune system somatic hypermutation | 5 exons of immunoglobulin determinants; specific for regulatory signals, not coding sequences52, 54 |
| Yeast retroviral-like elements Ty1-Ty4 | Strong preference for insertion upstream of RNA polymerase III initiation sites82 |
| Yeast retroviral-like element Ty1 | Preference for insertion upstream of RNA polymerase II initiation sites rather than exons83 |
| Yeast retroviral-like element Ty5 | Strong preference for insertion in transcriptionally silenced regions of the yeast genome84 |
| Drosophila P-factors | Preference for insertion into the 5 end of transcripts85 |
| Drosophila P-factors | Targeting to regions of transcription factor function by incorporation of cognate binding site86-89 |
| HeT-A and TART retrotransposons | Insertion at Drosophila telomeres90 |
| R1 and R2 LINE element retrotransposons | Insertion in arthropod ribosomal 28S coding sequences91 |
| Group I homing introns (DNA based) | Site-specific insertion into coding sequences in bacteria and eukaryotes92 |
| Group II homing introns (RNA based) | Site-specific insertion into coding sequences in bacteria and eukaryotes93 |
In other cases of targeting by the transcriptional control apparatus, the connection seems to be mediated by more transient factors (Table 3). For the yeast Ty3 retroviral-like element, which inserts with high reliability just upstream of sequences transcribed by RNA polymerase III, there has been a direct demonstration of interaction in vitro between virus-like particles and soluble PolIII transcription factors.96 One of the most intriguing examples is the targeting of P-factors, a class of DNA transposons involved in hybrid dysgenesis that are utilized as vectors for introducing exogenous sequences into the fruit fly genome.76 Incorporation of sequences containing transcription factor binding sites targets the newly constructed transposons to regions of the genome where those transcription factors operate with probabilities of about 30 50%.86-89 The targeting is not precise but regional (i.e. within a window of a few kilobase-pairs). Mechanistically, this indicates that DNA homology is not a component of targeting, which is probably based on protein-protein interactions of the bound transcription factor, as occurs in the guidance of RNA polymerase,
A 21ST CENTURY VIEW OF GENOME REFORMATTING IN EVOLUTION
Our knowledge of how natural genetic engineering functions and epigenetic control systems can reformat genomes is more than sufficient to support the evolutionary generalizations outlined in Table 1. We understand enough about genome organization and function and about natural genetic engineering to predict confidently that rapid episodes of major genome restructuring will become the focus of modern evolutionary theories. A great deal of attention will center on changes in the distribution of repetitive DNA elements and the profound phenotypic effects of such modifications to genome system architecture. Much of the creative aspects of genome reorganization are likely to involve "facultative" (i.e. non-essential and duplicated) components rather than coding and regulatory sequences directly involved in the maintenance of current phenotypes.97 Applying the information economy metaphor, we can think of these facultative components as constituting an R & D sector for the genome.98
The most profound, and most challenging, new aspect of thinking in a 21st Century fashion about evolution will be the application of information-processing ideas to the emergence of adaptive novelty. A major problem, often cited by religious and other critics of orthodox evolutionary theory, is how to explain the appearance of complex genomic systems encoding sophisticated multicomponent adaptive features.99, 100 The possibility that computational control of natural genetic engineering functions can provide an answer to the problems of Irreducible Complexity and Intelligent Design deserves to be explored fully. Contrary to the claims of some Creationists,99 these issues are not scientifically intractable. They require an application of lessons from the fields of artificial intellligence, self-adapting complex systems, and molecular cell biology.100, 101
We already have some clues about how to proceed in addressing complex novelties in evolution. As McClintock first demonstrated, insertions of MGEs at distinct genetic loci bring them under coordinate control.8, 64, 66 Thus, we know in principle how multi-locus genomic systems can originate. Once such systems exist, we know that the transcriptional regulatory apparatus is capable of specifically accessing the component loci in response to biologically meaningful signals. A number of observations now demonstrate that the transcriptional apparatus can also guide MGEs and other natural genetic engineering activities to the components of existing multi-locus systems (Table 5). Thus, at the molecular and cellular levels, it is plausible to postulate targeting of specific MGEs to dispersed genomic regions encoding a suite of interacting proteins. Such targeting can provide those proteins with a common new regulatory specificity or with shared novel activity domains. In this way, complex multi-locus systems can be adapted to new uses. Moreover, insertion of the same MGEs into previously unrelated genomic locations can recruit new molecular actors to build up new systems. This view is certainly consistent with the evidence from whole genome sequencing.33, 47
Naturally, most genome-wide natural genetic engineering experiments will not be adaptively useful and will be eliminated by natural selection. What is necessary for evolutionary success of organisms requiring new adaptations is that the process of heritable change be frequent enough and sufficiently biased towards the creation of functional systems that at least one experiment succeeds. On a truly random, one locus at a time basis, the probabilities are simply too small to have a chance of creating useful new multilocus systems within any realistic time frame.
A major virtue of this Symposium is the encounter of practicing scientists with philosophers and historians of science. That interdisciplinarity has allowed multiple levels of discourse and enlightened all participants on the connections between observations, theory and philosophical assumptions. The topic of evolution is one where these connections are deep, and the debates are particularly sharp. One philosophical question that has proved extraordinarily contentious concerns the respective roles of design and chance in evolution. This topic is so heated because it touches on fundamental differences between materialistic assumptions and religious faith. However, I argue that molecular discoveries about cellular information processing, epigenetic modifications of the genome, and natural genetic engineering place this issue in a new naturalistic perspective. We can now postulate a role for some kind of purposeful, informed cellular action in evolution without violating any tenets of contemporary science or invoking actors beyond experimental investigation. It remains to be established how "smart" cellular networks can be in guiding genome reformatting and sequence reorganization towards adaptive needs. Fortunately, the beginning of a new century finds us with the scientific tools and conceptual framework (Table 1) to ask questions whose answers may give us an entirely new vision of the fundamental properties of living organisms.
REFERENCES