A 21st Century View of evolution
James A. Shapiro
Department of Biochemistry and Molecular Biology,
University of Chicago, Chicago, IL 60637
ABSTRACT
Physicists question whether there are "universals" in biology. One reason is that the prevailing theory of biological evolution postulates a random walk to each new adaptation. In the last 50 years, molecular genetics has revealed features of DNA sequence organization, protein structure and cellular processes of genetic change that suggest evolution by natural genetic engineering. Genomes are hierarchically organized as systems assembled from DNA modules, which themselves generally constitute systems at lower levels. Each genome is formatted and integrated by sequence elements that do not code for proteins. These formatting elements constitute codons in multiple genetic codes for distinct functions such as transcription, replication, DNA compaction and genome distribution to daughter cells. Consequently, the genome has a computational system architecture. Proteins are systems composed of functionally distinct domains connected in polypeptide chains. Whole-genome sequencing indicates that rearrangement of genetic modules plus duplication and reuse of existing genomic systems are fundamental events in evolution. Studies of genetic change show that cells possess mobile genetic elements and other natural genetic engineering activities to carry out the necessary DNA reorganizations. Natural genetic engineering functions are sensitive to biological inputs, and their non-random operations help explain how novel system architectures can arise in evolution.
KEY WORDS: genome system architecture, signal
transduction, cellular computation, natural genetic engineering, mobile
genetic elements, repetitive DNA, DNA rearrangements
COMPLEXITY, GENETICS AND EVOLUTION: THE ARGUMENT IN A NUTSHELL
Our Symposium is entitled "Life as a Complex System." In order to understand this topic, we have to ask, "Why do living organisms use so many different molecules to carry out the basic tasks of survival, growth and reproduction?" With regard to genetics and evolution, I think an overall answer can be summarized in the following points:
Complexity permits sophisticated information processing. Cells have to deal with literally millions of biochemical reactions during each cell cycle and also with innumerable unpredictable contingencies. They are constantly evaluating multiple internal and external signals and adjusting their activities to continue the basic processes of survival and reproduction. Cells carry out their computations by a process of molecular interactions. More molecules means more powerful computational capacity.
Genomes integrate into cellular information processing because they are organized as computational storage organelles. That is, DNA serves as a data storage medium. To participate in cellular activities, genomes interact computationally with dynamic cellular complexes composed largely (but not exclusively) of proteins. As we shall see, genomes are built (Lego-like) of hierarchically organized modular systems. Much like the programs stored on a computer drive, genomic systems and subsystems are formatted by generic (i.e. repetitive) signals that provide functional addresses for the data in each module. The formatting is as important as the data (i.e. protein coding sequences) in providing a Genome System Architecture for each organism or species.
Evolutionary genomic change occurs largely by a process of Natural Genetic Engineering. Systemic genome organization means that new functions arise by the cut-and-splice rearrangement of genetic modules. Living cells possess mobile genetic elements and other biochemical functions which carry out the underlying DNA rearrangements. Cells regulate the activation of natural genetic engineering functions. Thus, cells have a capacity for major genome reorganization in response to evolutionary crisis. Moreover, the fact that natural genetic engineering changes are neither random in nature nor restricted to a single site in the genome means that they can create novel distributed (multilocus) systems and new genome system architectures.
UNIVERSALS IN BIOLOGY
At a meeting of physicists, it is important to address the issue of whether there are "Universals" in biology or, as many scientists believe, just many separate examples of specific systems, each evolved to take care of a particular adaptive need. One source of this latter view is the conventional theory that evolution occurs by a random walk through adaptive space and produces a virtually endless series of sui generis inventions. One alternative to this conventional view is that there exist design principles and procedures that are used repeatedly in evolution (in other words, evolution occurs as an engineering process). Consistent with this alternative "evolutionary engineering" view is the fact that there are, indeed, a number of Universals in biology (Table I).
Table I. Some Universals in Biology
Up to1953
The second post-1953 Universal, the recognition that the vast majority of genetic change results from the action of cellular biochemical systems that act on DNA, is far less widely known, and its significance is not appreciated outside a small group of specialists (3, 4). The discovery of built-in natural genetic engineering mechanisms dates back to Barbara McClintocks pioneering cytogenetic studies of the late 1940s and early 1950s (5). However, the ubiquity of internal systems for genome change only became apparent through molecular studies in bacteria in the 1960s and, with recombinant DNA technology, in eukaryotes in the 1970s and1980s (6-8). In terms of a 21st Century view of evolution, the major importance of natural genetic engineering is that this capability removes the process of genome restructuring from the stochastic realm of physical-chemical insults to DNA and replication accidents. Instead, cellular systems for DNA change, place the genetic basis for long-term evolutionary adaptation in the context of cell biology where it is subject to cellular control regimes and their computational capabilities (9-11).
HOW DOES THE GENOME FIT IN THE INFORMATION ECONOMY OF THE CELL?
The genome is the long-term storage medium for each species (much like a computer hard disk) and consists of the total information content of the DNA molecules in the cells of that species. Although most genomics researchers focus on the "coding" regions of the genome that determine the proteins a species can synthesize, genomes are built up of protein-coding and other classes of DNA sequences that are combinatorially formatted to carry out the multiple tasks necessary for overall genome function (Table II). While textbooks call the triplet code for amino acids in proteins "the genetic code," there are in fact many genetic codes for the different aspects of genome coding, packaging, replication, distribution, repair and evolution.
Table II. Different classes of genomic information
Coding sequences for RNA and protein molecules
Start and stop sites for transcription
Processing signals for primary transcripts
Control signals for level of expression
Control signals for dynamic access at right time
and place. Identifiers for coding sequences that must be coordinately or
sequentially expressed.
Signals for chromatin condensation/chromatin remodeling
Signals for DNA replication
Signals for distribution to daughter cells (non-random
chromosome partitioning)
Signals for error correction and damage repair
Signals for reprogramming when necessary
Specific references can be found in refs. 1, 2,
10, and 11.
An absolutely fundamental point to appreciate is that genomes only function through interaction with other dynamic information systems in the cell. By itself, DNA is inert. The information stored in DNA molecules requires interpretation by the highly dynamic cellular systems that control DNA packaging, imprinting, replication, transcription, translation, splicing, signal transduction, morphogenesis, and so forth. The significance of these essential interactions is that the genome necessarily constitutes part of a computational system in every living cell. The character of an organism is not determined solely by its genome. For example, in species with complex life-cycles, the same genome encodes two quite distinct organisms, such as the caterpillar and butterfly. We also see the cell-dependent nature of genome function in mammalian cloning, where a nucleus is removed from one terminally differentiated cell type that is capable only of a highly restricted range of genome expression and inserted into an egg cell, where the genome can provide the stored information for embryonic development and occasionally encode the formation of a normal individual (12, 13).
CELLULAR COMPUTATION AND GENOME SYSTEM ARCHITECTURE
There are many ways to visualize the systemic nature of genomic coding. One that is discussed in other parts of these PROCEEDINGS is the organization of protein molecules as systems of discrete structural and functional domains encoded in evolutionarily mobile DNA modules (14; these PROCEEDINGS, Symposia B6 and B7). Here we will examine the basic principles of gene expression and transcriptional regulation and the evolution of our one-dimensional concept of the gene into the more complex notion of a genetic locus.
The lac operon: A simple but illustrative example
Our example is the lac operon encoding the capacity for lactose utilization in the bacterium E. coli. Like virtually all classical genes, lac began existence as a point on a genetic map in the late 1940s, soon after the discovery of genetic exchange in bacteria (Fig. 1; see 15, 16, for detailed references to lac operon history).
![]()
Fig. 1. The lac gene, site of mutations affecting lactose utilization by E. coli (1947).
In the following years, Jacques Monod studied lac genetics because he had discovered that E. coli could discriminate between glucose and lactose; when given a mixture of the two sugars, the bacteria would invariably consume all the glucose before starting to consume the lactose. In 1961, Monod and his colleagues proposed the operon model. In the operon, lac had evolved into a system of "structural genes" encoding the proteins of lactose transport and metabolism (lacZYA), a "regulator gene" encoding a repressor (lacI), and a completely novel type of genetic element, the "operator" (lacO) (Fig. 2).
![]()
Fig. 2. The lac operon (1961)
It is important to recognize that lacO was not a "gene" encoding a product. Instead, it was a site on the DNA where the repressor bound to block the initial step of lacZYA expression from the adjacent DNA. This conceptual development was revolutionary in its impact on our understanding of genome function. Such cis-acting binding sites for proteins are now recognized as key components of the genome, essential for processes such as replication, transcription and genome distribution to daughter cells.
After the operon model, other scientists discovered additional binding sites in lac, including the site of RNA polymerase binding or "promoter" (lacP), a site for binding the Crp transcription factor that mediates glucose control of lacZYA transcription (CRP), and two additional operator sites that permit cooperative binding of the repressor (lacO2, lacO3). Thus, by the mid 1980s, molecular genetic analysis had decomposed the dimensionless lac gene into a structured system of protein-coding and cis-acting regulatory sites (Fig. 3).
![]()
Fig. 3. The lac operon (1990)
The importance of the organization of the various lac regulatory sites is that they permit the molecular computations that allow E. coli to discriminate glucose from lactose that is, to control expression of the lactose metabolic proteins so that they are only synthesized once glucose is no longer available. The basic biochemical reactions and molecular interactions involved in this computation can be stated as logical propositions that can then be combined into partial computations (Table III). These partial computations illustrate the molecular logic allowing the cell to execute the following overall computation: "IF lactose present AND glucose not present AND cell can synthesize active LacZ and LacY, THEN transcribe lacZYA from lacP."
Table III. Computational operations in lac operon regulation
Operations involving lac operon products
LacY + lactose(external) ==> lactose(internal)
(1)
LacZ + lactose ==> allolactose (minor product)
(2)
LacI + lacO ==> LacI-lacO
(repressor bound, lacP inaccessible) (3)
LacI + allolactose ==> LacI-allolactose
(repressor unbound,lacP accessible) (4)
Operations involving glucose transport components
and adenylate cyclase
IIAGlc-P + glucose(external) ==>
IIAGlc + glucose-6-P(internal) (5)
IIAGlc-P + adenylate cyclase(inactive)
==>
adenylate cylase(active) (6)
Adenylate cyclase(active) + ATP ==> cAMP
+ P~P (7)
Operations involving transcription factors
Crp + cAMP ==> Crp-cAMP (8)
Crp-cAMP + CRP ==> Crp-cAMP-CRP
(9)
RNA Pol + lacP ==> unstable complex
(10)
RNA Pol + lacP + Crp-cAMP-CRP ==>
stable transcription complex (11)
Partial computations
No lactose ==> lacP inaccessible
(3)
Lactose + LacZ(basal) + LacY(basal) ==>lacP
accessible
(1, 2, 4)
Glucose ==> low IIAGlc-P ==>
low cAMP ==> unstable transcription complex (5, 6, 7, 10)
No glucose ==> high IIAGlc-P
==>
high cAMP ==> stable transcription complex (5, 6, 7, 8, 9, 11)
Two aspects of this particular genomic computation deserve special mention. The first aspect is that the computation involves many molecules and compartments of the cell, not just DNA and DNA binding proteins. For example, the membrane transport proteins LacY and IIAGlc are essential. The second noteworthy aspect is that the computation involves the use of chemical symbolism as information is transmitted. Thus, the presence of allolactose inducer represents the availability of lactose and the ability of the cell to synthesize functional LacY and LacZ. Similarly, the concentrations of IIAGlc-P and cAMP represent the availability of glucose to the cell. Both whole cell involvement and transient chemical symbols are typical of cellular computation and signal transduction in general.
Organization of higher order systems in the genome
The lac operon is a relatively simple and thoroughly studied example of a genomic system. Despite its simplicity, it illustrates the basic features of how the genome is organized for interaction with other cellular information systems. One way to build higher-level systems in the genome is to use common binding sites at multiple genetic loci (10, 11, 17, 18 and U. Alon, these PROCEEDINGS). The CRP site serves in just this way. It is located in the transcriptional regulatory regions of diverse loci in the E. coli genome, connecting them into a network of functions regulated by the availability of glucose as a preferred growth substrate (15).
Another complementary way of building up higher-order systems is to complexify the structure of the transcriptional regulatory regions in genetic loci so that they contains many more protein binding sites. This strategy has been adopted in higher eukaryotes that undergo elaborate processes of cellular differentiation and multicellular development. Elaborate transcriptional regulatory architectures permit the formation of developmental expression networks in which multiple genetic loci are coordinately regulated by shared subsets of specific binding site motifs (19). Moreover, the combinatorial diversity of complex regulatory regions means that the expression of each genetic locus is subject to computationally intricate controls as the intracellular concentrations of different transcription factors change. A particularly well-studied example of such a regulatory region in the sea urchin has been analyzed at both the molecular and computational levels (20).
The transcription factor binding sites that serve to integrate multilocus genetic systems are one class of the highly diverse family of repetitive DNA sequences. With some exceptions, repetitive sequences do not encode proteins, and their occurrence at multiple locations in the genome distinguish them from classical genetic loci that map to specific sites. These repetitive sequences are major players in genome organization. In the draft human genome, for example, only 3% of the sequence information is in protein-coding exons, but over 45% is in repeated DNA sequences (21,22). Repetitive DNA is far more taxonomically specific than protein-coding DNA and serves as the most reliable indicator of identity for a species or even, as used in forensic analysis, for identifying an individual (23).
Contrary to what most people believe (and to what is often asserted in discussions of genome evolution), the fact is that repetitive DNA elements play critical roles in functional organization of the genome (Table IV). The effects on gene expression are quite dramatic and relate to the role of repetitive DNA in altering the structure of DNA compacted with histones and other DNA binding proteins into an overal structure called "chromatin." Repetitive DNA tends to form a densely packed DNA-protein complex called "heterochromatin" which generally inhibits both expression and replication of the underlying DNA sequences. Classical cytogenetic studies of a phenomenon called "position effect" have shown that expression of many genetic loci can be dramatically inhibited by proximity to heterochromatic regions of the genome (10, 24, 25). By forming extended domains of distinct chromatin configuration, the distribution of repetitive DNA elements can coordinately regulate expression of groups of linked genetic loci.
Table IV.Functions of repetitive DNA sequence elements (refs. in 10, 11)
Coordinated expression of unlinked genetic loci:
activator regions, silencing regions
DNA replication origins and chromosome end stability
(telomeres)
Chromosome distribution during cell division:
centromeres, chromosome pairing
Chromatin organization and gene expression in
development: chromatin domains, heterochromatin nucleation, insulator elements,
position effects
The above considerations and many other studies too numerous to mention here have led to the concept that genomes have an overall organization (Table V). In keeping with our computational analogy, we called this overall organization, unique to each species, its Genome System Architecture.
Table V. Genome System Architecture
Genome as an information storage organelle (cf.
disk drive)
Formatting for coding sequence expression: translation,
codon bias, splicing, etc.
Formatting for transcription: regulatory regions
(5, 3, and intron), chromatin domains
Formatting for replication: origins, telomeres,
chromatin domains
Formatting for DNA condensation and spatial organization
in the nucleus: euchromatin, heterochromatin, laminar attachment sites
Formatting for genome segregation: centromeres,
chiasmata
Formatting for genome reorganization and natural
genetic engineering: transposable and repetitive elements, DNA rearrangement
signals
==> "Systems all the way down" (organized
by repetitive signals)
IMPLICATIONS OF GENOME SYSTEM ARCHITECTURE FOR EVOLUTIONARY CHANGE
If it is true, as all sequencing projects indicate, that genomes are composed of Lego-like assemblies of smaller and larger modular genetic elements (segments of protein coding sequences, regulatory sites, repetitive DNA elements, chromatin domains), then it follows that a major source of genetic novelty must be the rearrangement of these modular components. For example, a new protein function can be generated by the rearrangement of existing coding modules (often, referred to as "domain- or exon-swapping," 26). In addition, patterns of gene expression and coordination of multiple genetic loci can arise through the distribution of transcriptional regulatory sites among new groups of genetic loci or by rearranging the combinations of binding sites in complex control regions. Even higher order changes in functional organization can occur by creating new combinations of linked genetic loci and/or altering the boundaries of chromatin domains. This last process can occur through redistributing repetitive DNA elements without changes in the structure of individual genetic loci. In other words, by a process of cut-and-splice genetic engineering leading to the appropriate changes in the arrangements of diverse genetic modules, genomes can be altered locally, regionally or globally, and they can be reformatted to acquire distinct system architectures. It may be postulated that such reformattings are more important events in the origin of new species and genera than gradual changes in proteins.
The existing whole genome sequences (in particular, the draft human genome sequence) appear to support this view of genome evolution. There is a pattern of the retention and reuse of genomic systems, which can often be adapted to novel cellular or organismal functions. The following general features have been observed:
Functional modules tend to be amplified; these modules can be domains, entire protein determinants, regulatory sites, entire genetic loci, complex higher order assemblies of multiple genetic loci, such as the homeobox clusters (27), or long chromosome segments (e.g.28). The importance of whole or partial genome duplications in evolution was pointed out many years ago by Ohno (29).
Multiple chromosome rearrangements are involved in the relocation of amplified modules throughout the genome or in reorganizing similar chromosome segments shared between related organisms (21, 30, 31).
There are major differences in the identity, numbers and locations of repetitive sequence elements between related organisms; often these repetitive elements define specific functional regions, such as the tandem arrays that help define chromosome centromeres (e.g. 28).
WHERE DOES GENETIC CHANGE REALLY COME FROM?
The conventional view is that genetic change comes from stochastic, accidental sources: radiation, chemical, or oxidative damage, chemical instabilities in the DNA, or from inevitable errors in the replication process. However, the fact is that DNA proofreading and repair systems are remarkably effective at removing these non-biological sources of mutation. For example, consider that the E. coli cell replicates its 4.6 megabase genome every 40 minutes. That is a replication frequency of almost 2 kHz. Yet, due to the action of error-recognition and correction systems in the replication machine and in the cell to catch mistakes in already-replicated DNA, the error rate is reduced below one mistake in every 1010 base-pairs duplicated, and a similar low value is observed in mammalian cells (32). That is less than one base change in every 2000 cells, certainly well below the mutation frequencies I have measured in E. coli of about four mutations per every 100 to 1000 cells.
In addition to proofreading systems, cells have a wide variety of repair systems to prevent or correct DNA damage from agents that include superoxides, alkylating chemicals and irradiation (33). Some of these repair systems encode mutator DNA polymerases which are clearly the source of DNA damage-induced mutations and also appear to be the source of so-called "spontaneous" mutations that appear in the absence of an obvious source of DNA damage (34). Results illustrating the effectiveness of cellular systems for genome repair and the essential role of enzymes in mutagenesis emphasize the importance of McClintocks revolutionary discovery of internal systems generating genome, particularly when an organism has been challenged by a stress affecting genome function (Fig. 4; 5).
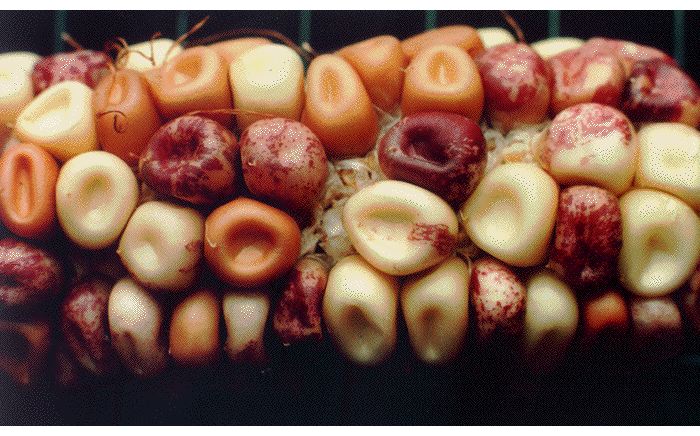
Fig. 4. A maize ear illustrating the kind of internally-generated genetic changes studied by McClintock.
McClintock recognized that genetic change is a cellular process, subject to regulation, and is not dependent on stochastic accidents. The idea of internally-generated, biologically regulated mutation has profound impacts for thinking about the process of evolution. Darwin himself acknowledged this point in later editions of Origin of Species, where he wrote about natural "sports" or "...variations which seem to us in our ignorance to arise spontaneously. It appears that I formerly underrated the frequency and value of these latter forms of variation, as leading to permanent modifications of structure independently of natural selection." (6th edition, Chapter XV, p. 395).
To see the real-world evolutionary importance of built-in biological mechanisms of genetic change, we have only to consider the post-WWII emergence of multiple antibiotic resistance in bacteria. This phenomenon represents the largest and best-documented evolutionary experiment in the molecular biology era. Interestingly, when antibiotic use began, we had a robust theory of how resistance would evolve by modification of existing cell components so that they were no longer antibiotic-sensitive. This theory was confirmed by laboratory experiments. Nonetheless, when the basis of naturally evolving multiple antibiotic resistance was determined, the experimentally-confirmed theory was wrong. Resistance resulted from the presence of new biochemical activities in the bacteria, encoded by new transmissible genetic systems that could accumulate additional DNA encoding these resistance activities (35).
NATURAL GENETIC ENGINEERING
Mobile Genetic Elements
As might be expected from the patterns of genetic reorganization observed in whole genome databases, living cells possess a variety of biochemical systems capable of creating novel DNA structures. Sometimes these systems are individual proteins and create localized changes, such as the mutator DNA polymerases mentioned above, or the site-specific recombinases that join together rare recognition sites (site-specific recombination; 9, 35). Sometimes, much larger multiprotein assemblages are involved, like the apparatus for carrying out homologous genetic recombination or for repairing severed DNA molecules by non-homologous joining of broken ends (36). Among the most important systems are those called "mobile genetic elements" (MGEs; 7, 8), which make up about 43% of the human genome (21). These MGEs include the transposable "controlling elements" discovered by McClintock, and they comprise integrated systems of proteins and nucleic acids that interact to mobilize DNA to new locations in the genome.
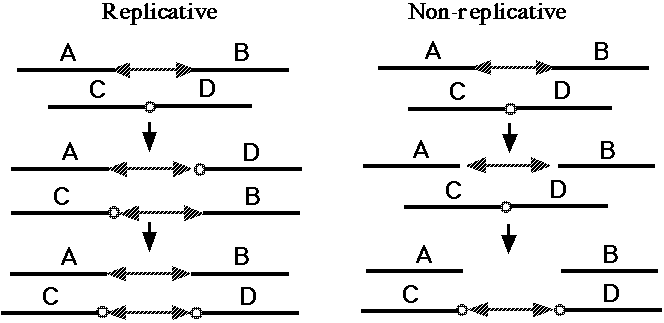
Fig. 5. Replicative and non-replicative mechanisms of DNA transposition (37). The double-headed arrows indicate a DNA transposon. The small circles indicate a short sequence at the target site; the duplication of this target sequence is a hallmark of almost all transposition events [37, 38].
One broad class of MGEs are those that act on the genome purely at the DNA level. The MGEs that mobilize antibiotic resistance determinants are DNA-based elements called "transposons," and this name has become general for all MGEs that operate at the DNA level. Both prokaryotes and eukaryotes possess transposons that have two basic activities. They can mobilize themselves to new sites in the genome by one of two related mechanisms (Fig. 5, 37), and they can rearrange adjacent segments of the genome in a variety of characteristic ways (Fig. 6, 38). What is of basic importance here is the fact that transposons can make any segment of the genome mobile, and thus capable of duplication, and that they can generate precisely the type of large-scale rearrangements observed in sequenced genomes. There are about 300,000 DNA transposons in the human genome (3% of the genome).
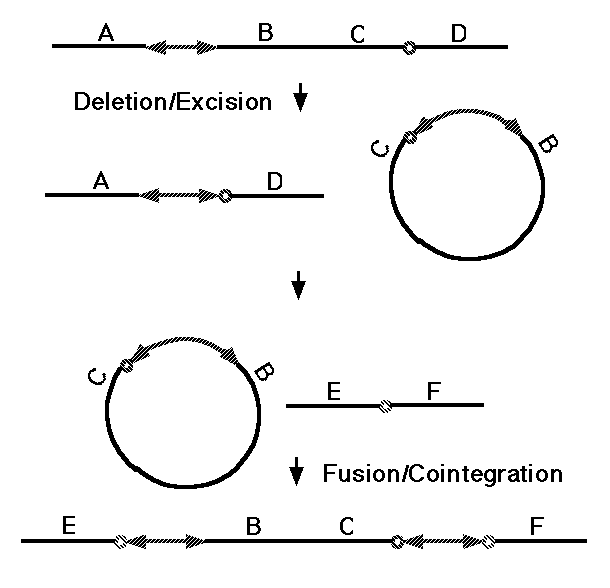
Figure 6. Transposition of a chromosomal segment
to a new site in the genome mediated by a replicative transposon (see 38
for details).
In eukaryotes, there are additional classes of MGEs based on the reverse transcription of RNA into DNA copies which can then be inserted into new genomic locations. These elements are called "retrotransposons," and they come in two basic varieties (39). One variety includes retroviruses, such as HIV, whose DNA proviruses can move from the genome of one cell to that of another via infectious particles, and non-infectious retroviral-like elements whose DNA can move from one site to another within the same nucleus. The integrated DNA structures of this retroviral class of retrotransposons are characterized by long terminal repeat or LTR structures at each end. The human genome contains about 450,000 LTR retrotransposons (8% of total DNA). A second class of retrotransposons consists of elements lacking terminal repeats but having a polyA tail at one end, like a synthetic cDNA. These polyA-tailed MGEs fall into two major classes: LINEs, or Long Interspersed Nucleotide Elements, and SINES, or Short Interspersed Nucleotide Elements. There are, respectively, 850,000 and 1,500,000 LINEs and SINEs in the human genome (representing 21% and 13% of total DNA).
Like LTR retrotransposons, LINE elements encode their own reverse transcriptase and integration activities. Thus, once the original MGE has been transcribed, both classes can copy the RNA into DNA and reinsert the DNA copy at a new location. The SINE elements do not encode reverse transcriptase activity and appear to depend upon LINEs for the copying and chromosomal integration of their transcripts. It is quite significant that the unusually abundant SINE elements are often highly specific for particular taxonomic groups. For example, a number of mammalian orders (primates, rodents, artiodactyls, etc.) share LINE and some less abundant SINE elements, but the most abundant SINEs in each orders genome are limited to that order (40). Thus, a primate cell can be distinguished from a rodent cell simply by examining the SINEs, and genetic changes involving these taxonomically-limited SINEs are unique to the group which possesses them.
Retrotransposons are powerful agents of genome restructuring. Because they are so abundant, they can mediate large scale chromosome restructurings by means of homologous recombination between similar elements at distant locations. However, they have other unique capabilities. The LTRs of the retroviral class contain potent signals for initiating and terminating transcription. Thus, their insertion near or within a genetic locus can place it under novel transcriptional controls, as happens when leukemia viruses activate cellular oncogenes. Some SINE elements also have transcription factor binding sites in their sequences and can place adjacent coding sequences under specific new kinds of regulation (41, 42). Moreover, insertion of SINEs into introns can create novel exons and add sequence motifs to pre-existing proteins (43). In some cases, the coding sequences for particular proteins are largely derived from SINEs and create functional molecules that can be found only in the originating taxon (43).
LINEs are master genetic engineers, and it has been argued that they are the major force in structuring mammalian genomes (44). Often LINE transcription does not terminate at its polyA tail but reads through into adjacent DNA; upon reverse transcription and integration into an intron, this adjacent sequence material can form part of a new regulatory region or a coding exon. In fact, LINE-mediated exon shuffling to create new multidomain proteins has been experimentally demonstrated (45). In addition, LINE element activities can reverse transcribe and insert cellular mRNAs into the genome, creating extra intron-free copies of a coding sequence. This process is the source of "processed pseudogenes" in the genome, and apparently it also has played a major role in the amplification of olfactory receptor proteins that are major adaptive inventions of mammals (46).
Non-randomness and Regulation of Natural Genetic Engineering Activities
The foregoing discussion and an extensiveliterature that cannot be cited here make it clear that MGEs and other natural genetic engineering functions have the capacity to reorganize genomes in just the ways needed to reformat modular genome system architectures. This point is increasingly recognized (e.g. 21,22). However, the degree to which these genome reorganization activities are not random is poorly appreciated. Non-randomness is evident at three levels: mechanism, timing, and sites of action.
Mechanistically, we can appreciate the very characteristic ways that MGEs rearrange DNA from schemes like Fig. 6. The ability to create large segments flanked by copies of the MGE is built into the process of replicative transposition (38). Another kind of non-randomness occurs when a proretrovirus inserts in a new genome location. In that case, a whole package of transcriptional and other regulatory signals in the LTRs has been placed next to new sequences, creating the potential for a new genomic system. The action of LTR-containing retrotransposons in yeast (and also certain transposons in bacteria) as mobile activator elements illustrates the functional utility of such "package deal" rearrangements (7, 9).
As far as timing of natural genetic engineering is concerned, McClintock emphasized the importance of stress events she called "genome shocks" for activating the built-in systems of DNA rearrangement. Now that these systems have been investigated at the molecular level, we have many examples illustrating how natural genetic engineering can be kept latent during normal proliferation but specifically activated in response to particular signals (see 9, 10, 16 for specific references not given below).
In specialized DNA rearrangement systems, like the ones used in our immune cells to generate an enormous variety of coding sequences for antibodies and T-cell receptors, the necessary genetic engineering functions turn on in response to developmental controls.
Similarly, a yeast retrotransposon undergoes transcription, reverse transcription and integration in response to mating pheromone.
In bacteria, the phenomenon of "adaptive mutation" occurs when cells activate MGEs and mutator polymerases in response to long-term starvation signals (16, 47).
A particularly important source of rapidly-activated natural genetic engineering called "hybrid dysgenesis" takes place when individuals mate with individuals from a distinct interbreeding group or species (7). Hybrid dysgenesis results in extraordinarily high rates of mutation and chromosome rearrangements caused by DNA transposons and LINE elements in fruit flies, DNA transposons in nematode worms, and retroviruses in mice and wallabies (48).
In addition to temporal specificity, it turns out that many natural genetic engineering functions show intriguing degrees of selectivity in where they act within the genome. This selectivity appears to be chiefly related to interactions between natural genetic engineering systems and the cellular systems controlling transcription and chromatin formatting. The examples we have of target selection include the action of localized point mutagenesis, retrotransposons and DNA transposons (see 9, 10 for specific references):
50-75% preference for insertion of yeast retrotransposons Ty1-4 upstream of RNA polymerase III transcription start sites; with Ty3, a direct interaction has been demonstrated between the integration complex and PolIII transcription factors.
Preference of Ty1 insertion upstream of RNA polymerase II start sites.
Preference for Ty5 retrotransposon insertion into transcriptionally silenced regions of the yeast genome.
Preference for P factor insertion into the 5 end of transcripts (50).
Targeting of genetically engineered P factor vectors containing transcription factor binding sites to short chromosome regions known to bind the corresponding factor.
Specificity of the HeT-A and TART retrotransposons for chromosome ends in fruit flies.
A 21ST CENTURY VIEW OF EVOLUTION
Evolution is the history of organisms that have succeeded in adapting to changing circumstances. Over evolutionary time, this means altering the genome the long-term information storage organelle of all living cells to provide the functional information needed to survive and reproduce in new conditions. Those organisms that have the most flexible computational capabilities, in particular those that have the best means of altering information stored in the genome, will have an advantage. Thus, it makes sense for organisms to possess crisis-responsive natural genetic engineering functions, and we should not be surprised to find them ubiquitous in contemporary organisms, all of whom are evolutionary winners. Indeed, it is now difficult to imagine how organisms that depend upon gradual accumulation of stochastic mutations could persist in the evolutionary rat race.
The last half century has taught us an astonishing amount about how living organisms function at the molecular level, in particular about how they execute cellular computations through molecular interactions and about the systemic, modular, computation-ready organization of the genome. We have come to realize some of the basic design features that govern genome structure. Combining this knowledge with our understanding of how natural genetic engineering operates, it is possible to formulate the outlines of a new 21st Century vision of evolutionary engineering that postulates a more regular principle-based process of change than the gradual random walk of 19th and 20th Century theories. Such a new vision is not all-encompassing because it cannot provide detailed accounts for major events currently beyond the reach of science, such as the origin of cellular life or the mechanisms of endosymbiotic events underlying the emergence of distinct superkingdoms and kingdoms of life (51, 52). Nonetheless, a 21st Century view of evolution can help us understand how new taxonomic groups have emerged bearing novel complex adaptations.
As I see it, a 21st Century view of evolution has to include the following features:
Major alterations in the content and distribution of repetitive DNA elements results in a reformatting of the genome to function in novel ways --without major alterations of protein coding sequences. These reformattings would be particularly important in adaptive radiations within taxonomic groups that use the same basic materials to make a wide variety of morphologically distinct species (e.g. birds and mammals).
Large-scale genome-wide reorganizations occur rapidly (potentially within a single generation) following activation of natural genetic engineering systems in response to a major evolutionary challenge. The cellular regulation of natural genetic engineering automatically imposes a punctuated tempo on the process of evolutionary change.
Targeting of natural genetic engineering processes by cellular control networks to particular regions of the genome enhances the probability of generating useful new multi-locus systems. (Exactly how far the computational capacity of cells can influence complex genome rearrangements needs to be investigated. This area also holds promise for powerful new biotechnologies.)
Natural selection following genome reorganization eliminates the misfits whose new genetic structures are non-functional. In this sense, natural selection plays an essentially negative role, as postulated by many early thinkers about evolution (e.g. 53). Once organisms with functional new genomes appear, however, natural selection may play a positive role in fine-tuning novel genetic systems by the kind of micro-evolutionary processes currently studied in the laboratory.
Molecular
genetics has amply confirmed McClintocks discovery that living organisms
actively reorganize their genomes (5). It has also supported her view that
the genome can "sense danger" and respond accordingly (56). The recognition
of the fundamentally biological nature of genetic change and of
cellular potentials for information processing frees our thinking about
evolution. In particular, our conceptual formulations are no longer dependent
on the operation of stochastic processes. Thus, we can now envision a role
for computational inputs and adaptive feedbacks into the evolution of life
as a complex system. Indeed, it is possible that we will eventually see
such information-processing capabilities as essential to life itself.
REFERENCES